
The following posters will be presented at the Poster Session on Monday 9th March at 18:30 – 19:30. The poster prizes have been sponsored by IKTOS.
Industrialising X-Ray Fragment Hit Progression – Anthony Aimon (Diamond Light Source Ltd)
XChem(1) is high-throughput fragment screening facility set up by Frank von Delft at the national synchrotron Diamond Light Source that uses the X rays to identify fragments binding to proteins of high biological relevance (cancer treatment, bacteria resistance etc.). Since 2015 it has hosted over 100 academic and industry experiments, measuring >200,000 crystals and yielding >2000 fragment hits. For one experiment, up to 1000 crystals can be soaked and mounted, the data collected in the beamline and analysed in one week. However, the real impact of this work will only become apparent when we manage to routinely progress numerous weakly binding fragments to drug-like compounds. We have decided to tackle this by: 1) developing a set of computational tools for the design of follow-up compounds (https://fragalysis.diamond.ac.uk/, under development); 2) building an automated chemistry platform that can quickly provide 100s of compounds, necessary for the progression of the many potential vectors per fragment. In this poster we will focus on the set-up of the high-throughput synthetic platform with cheap robotics and the chemical reactions that have been successfully automated (single- and multi-step) to synthesise a high number of diverse fragment follow-ups. Secondly we demonstrate that crude reaction mixtures can be directly used in X-ray screening experiments which removes one of the biggest hurdle in current automated chemistry processes.
(1) – https://www.diamond.ac.uk/Instruments/Mx/Fragment-Screening.html
Reaction InChI (RInChI): Present and Future – Gerd Blanke (StructurePendium Technologies GmbH)
The International Chemical Identifier for Reactions (RInChI) provides a vendor-neutral, machine-readable string representing chemical reactions. The prototype of RInChI was first released in 2011 and Version 1.00 has been downloadable since 2017. RInChI is starting to be used in databases and cheminformatics software packages, drawing tools are beginning to provide the calculations of RInChIs from reaction depictions and publishers are going ahead with integrating the RInChI into their web pages. We will discuss what has been achieved thus far and plans for the future. This will include adding data to auxiliary information layers, including the ability to assign atom-atom mapping information and chemical process details, such as reaction temperature and yield. Information like this can be used to optimize reaction pathways leading to high levels of automation of smart chemical syntheses. The planned extensions will incorporate future developments of the InChI standard.
Artificial Labels for Multi-label Training of Neural Networks Policies for Retro-Synthetic Template Selection and Route Finding – Esben Jannik Bjerrum (AstraZeneca)
Computer assisted retrosynthetic planning and route-finding algorithms are useful tools in chemistry. Research into algorithms and approaches has seen renewed interest with the emergence of deep learning enhanced approaches. Deep learning can significantly speed up the search for routes in tree search algorithms1 or directly suggest retro-synthetic disconnections in template-free approaches2,3. We have implemented a neural network guided tree search algorithm for retrosynthetic planning inspired by the 3N-MCTS algorithm1 and deployed it for project use in the medicinal chemistry labs via a web-based front-end. We are further investigating how it can be used as part of our de novo design engine REINVENT4. Further development of the retro-synthetic algorithm has focused on the performance of the neural network that selects and prioritizes retrosynthetic templates from large template libraries extracted from reaction databases. We first investigated the influence of both commercial and public data sources for template coverage and their influence on route finding capabilities. The accuracy of template re-finding for the policy network was found to be a bad predictor of the final route-finding capabilities of the network. The accuracy of re-finding of a single template can be misleading, as a given product can be the result of several different connections 5. After receiving feedback from chemist about issues with breaking down ring systems and heterocycles, we have further investigated how policy networks trained on a subset of the reaction space, here ring-forming reactions, can lead to improved performance for the specific sub-task enabling targeting retro-synthetic analysis of ring systems 6. Moreover, the current way of training template selection and prioritization policy networks leads to a lot of false positives in the top-50 predictions. These are reaction templates that can’t be applied in silico to the given product and need to be filtered away post-prediction, leading to a more inefficient route search. By using artificially created labels and post-training modifications to the policy networks, we can significantly lower the amount of non-applicable templates in the top-50 predicted templates and ultimately influence the route-finding capability of the algorithm.
(1) Segler, M. H. S.; Preuss, M.; Waller, M. P. Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI. Nature 2018, 555 (7698), 604–610. https://doi.org/10.1038/nature25978 .
(2) Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3 (10), 1103–1113. https://doi.org/10.1021/acscentsci.7b00303 .
(3) Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V. H.; Haeuselmann, R. A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Predicting Retrosynthetic Pathways Using a Combined Linguistic Model and Hyper-Graph Exploration Strategy. 2019, 1–26 http://arxiv.org/abs/1910.08036 .
(4) Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular De-Novo Design through Deep Reinforcement Learning. J. Cheminform. 2017. https://doi.org/10.1186/s13321-017-0235-x .
(5) Thakkar, A.; Kogej, T.; Reymond, J.-L.; Engkvist, O.; Bjerrum, E. J. Datasets and Their Influence on the Development of Computer Assisted Synthesis Planning Tools in the Pharmaceutical Domain. Chem. Sci. 2019. https://doi.org/10.1039/C9SC04944D .
(6) Thakkar, A.; Selmi, N.; Reymond, J.-L.; Engkvist, O.; Bjerrum, E. J. ‘Ring Breaker’: Assessing Synthetic Accessibility of the Ring System Chemical Space. 2019. https://doi.org/10.26434/chemrxiv.9938969.v2 .
Development of an automated gas phase catalytic microreactor platform for kinetic studies – Solomon Gajere Bawa (University College London)
Flow microreactors have been proven as an effective experimental tool due to superior mass and heat transfer, improved safety and low material consumption. As such, they enable isothermal kinetic studies with minimal material resources. Ease of automation and integration with online analysis allows a large amount of experimental data to be generated rapidly. An automated system based on a flow micropacked bed catalytic reactor was developed in this work to conduct pre-planned experiments. The microreactor was fabricated using photolithography and deep reactive ion etching of a silicon wafer. Anodic bonding was used for sealing the silicon to a glass cover. Isothermal operation was possible owing to the microreactor high surface to volume ratio. The main microreactor channel width was 2 mm and depth 0.42 mm. Serpentine channel between the microreactor inlet and the main channel aided the mixing of the gaseous feed. Methane combustion on Pd/Al2O3 catalyst was selected as reaction to test the performance of the developed automated system. The feed consisted of 5% CH4/He, O2 and N2 as internal standard. Online Gas Chromatography (GC) was used to perform analysis of the outlet stream. The experimental system was monitored and controlled by LabVIEW. Computation of GC results was done by a Python script that communicated with LabVIEW through the Python integration toolkit. Safety measures with respect to temperature and pressure were included in the automated platform. Temperature difference was found to be < 3⁰C at operating temperature of 350⁰C across the packed bed. The reproducibility of the system was ascertain with relative standard deviation of 2% at higher conversion (74-94%), and 10% at the lowest conversion (6%). Difference in carbon balance of reactant and product was < 3% within the experimental conditions investigated. Methane conversion observed at 300⁰C was 22.3 and 22.5% for a reaction time of 40 and 200 minutes respectively, which inferred good catalyst stability. Within each experimental campaign, the system adjusted the process variables (methane concentration, oxygen-methane ratio, temperature and total flow rate) and collect information on the experimental composition of the product stream (via GC) in a continuous mode, when running a set of factorial experiments without user intervention. The platform will be used for kinetic model identification to discriminate among possible models and estimate the set of kinetic parameters of each potential model.
AI/Machine Learning for Chemical Development – David Buttar (AstraZeneca)
AstraZeneca is developing a platform for data mining coupled with ML/AI to exploit in developing robust, scalable chemical processes for drug development. It is believed that the integration of process focused reactivity data and machine learning will drive the development of novel predictive process models. Such predictive models can be utilised by development chemists to rationalise and improve route selection activities, with the goal of faster, more efficient and sustainable drug development. The poster describes the targets and challenges for development chemistry and the vision for a next generation AI/ML chemical development platform.
Active Learning for Cost-Efficient Reaction Prediction using Kinetic Data – Paul Dingwall (Queen’s University Belfast)
Predicting the performance of a reaction is a challenging task. The reports that do so typically use machine learning algorithms that are fed a large volume of single timepoint yield (STPY) data, generated via high throughput experimentation, and an even larger volume of computed molecular descriptors to create predictive models. The use of STPYs to describe the outcome of catalytic reactions is a major experimental shortcoming of existing work. If a reaction only reached 50% would it reach 99% if run for longer? How much longer? Was there a slow catalysts activation period followed by a rapid reaction? Or was a catalyst poisoned, stopping the reaction midway? These are factors described by the reaction kinetics. Kinetic data should outperform STPY data in predicting the performance of homogeneously catalysed reactions. Rather than simply predicting a numerical value for yield, as is the case with STPYs, by predicting a kinetic profile, the reaction yield or conversion can be calculated for a given reaction time. Machine learning techniques applied in existing work using STPYs will not be appropriate for handling kinetic profiles; these data represent a trajectory of time-correlated points rather than a single stationary point. The goal of this project is to use machine learning trained on kinetic data to predict entirely new kinetic profiles. An experimental hurdle exists in that collecting kinetic data is a slow and laborious process, meaning both low data volumes and throughput. Active learning represents an optimal and highly cost-effective machine learning approach in this context.
Catalyst Seeking Substrate: Computational Prediction in Homogeneous Organometallic Catalysis – Derek James Durand (University of Bristol)
Derek J. Durand1, Dr. Alex Cresswell2, Dr. Natalie Fey1, Dr. Jason Lynam3, Prof. Paul Pringle1, Dr. Ruth Webster2.
1) School of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS, UK.
2) Department of Chemistry, University of Bath, Soldier Down Ln, Bath, BA2 7AX, UK.
3) Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK.
The computational optimisation and design of homogeneous transition-metal catalysts using mechanistic studies based on density functional theory (DFT) calculations has been demonstrated extensively. Such studies can be enhanced by ‘chemical intuition’ and molecular descriptor-based predictive modelling.1 Calculated ligand descriptor databases, such as Bristol’s Ligand Knowledge Bases,4,5 have provided a foundation to explore chemical ligand space and identify reactivity ‘hotspots’ for catalyst optimisation. However, such models are dependent on suitable experimental training data, making them reaction specific.6 Recent reports highlight descriptors and models which better account for the nature of the catalyst during reaction rather than isolation to build useful predictive models for reaction performance.3, 7 The development of a dual-purpose reactivity descriptor approach, suitable for both in-silico catalyst screening and predictive modelling of wider reactivity space is therefore highly desirable. This contribution describes the initial development of the Bristol Reactivity in Catalysis Knowledge-base (BRiCK), which aims to fulfil this goal. Utilising well-known cationic rhodium complexes bearing bisphosphine ligands as model catalysts,8, 9 the development of a standard computational workflow for exploring additions to multiple bonds is presented, along with results for a representative sample of calculations, covering a range of hydrofunctionalisation reactions.
- S. Ahn, M. Hong, M. Sundararajan, D. H. Ess and M.-H. Baik, Chem. Rev., 2019, 119, 6509-6560.
- D.-H. Kwon, J. T. Fuller, U. J. Kilgore, O. L. Sydora, S. M. Bischof and D. H. Ess, ACS Catal, 2018, 8, 1138-1142.
- G. Skoraczyński, P. Dittwald, B. Miasojedow, S. Szymkuć, E. P. Gajewska, B. A. Grzybowski and A. Gambin, Sci. Rep., 2017, 7, 3582.
- D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561-6594.
- J. Jover, N. Fey, J. N. Harvey, G. C. Lloyd-Jones, A. G. Orpen, G. J. J. Owen-Smith, P. Murray, D. R. J. Hose, R. Osborne and M. Purdie, Organometallics, 2012, 31, 5302-5306.
- N. Fey, S. Papadouli, P. G. Pringle, A. Ficks, J. T. Fleming, L. J. Higham, J. F. Wallis, D. Carmichael, N. Mézailles and C. Müller, Phosphorus, Sulfur, Silicon Relat. Elem. 2015, 190, 706-714.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631.
- R. R. Schrock and J. A. Osborn, J. Am. Chem. Soc., 1976, 98, 2134-2143.
- J. A. Osborn and R. R. Schrock, J. Am. Chem. Soc., 1971, 93, 2397-2407.
RetroBiocat: a tool for computer aided synthesis planning of biocatalytic cascades – William Finnigan (University of Manchester)
William Finnigan, Lorna Hepworth, Sabine L. Flitsch & Nicholas J. Turner.
Computer aided synthesis planning (CASP) seeks to augment the abilities of chemists or biologists for the design of pathways to a molecule of interest. Recently, a number of tools have been shown to make useful suggestions for the design of routes for chemical syntheses, or for new metabolic pathways in microbial hosts 1,2. Despite these recent successes, CASP tools for the design of synthetic biocatalytic cascades remain under-developed, in spite of the advantages biocatalysis can offer for organic synthesis. Here we present RetroBiocat, a tool for computer aided biocatalytic retrosynthesis, available as both a web-app and a python library. Using a set of manually curated reactions rules to describe biotransformations of industrial relevance 3, and a database containing information on the substrate specificity of some of these enzymes, we show our tool to be capable of identifying promising biocatalytic pathways to target molecules. We build upon CASP tools for both chemistry and synthetic biology, utilising knowledge of what synthetically useful reactions are, and which enzymes can reliably be used implement them. We demonstrate two modes, one for human-led exploration of a network of potential biotransformations, and another for generating automated pathway suggestions. To illustrate the application of our tool, we provide a number of example use-cases for the synthesis of industrially relevant target molecules.
- Delépine, B., Duigou, T., Carbonell, P. & Faulon, J. L. RetroPath2.0: A retrosynthesis workflow for metabolic engineers. Metab. Eng. 45, 158–170 (2018).
- Coley, C. W., Rogers, L., Green, W. H. & Jensen, K. F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 3, 1237–1245 (2017).
- Turner, N. J. & Humphreys, L. Biocatalysis in organic synthesis: The retrosynthesis approach. (Royal Society of Chemistry, 2018).
Predicting Cytochrome P450 Sites of Metabolism – Elena Gelžinytė (University of Cambridge)
Cytochromes P450 are a family of enzymes that are responsible for metabolising around 90% of pharmaceutical compounds in humans. Therefore, they are necessary to consider in drug discovery campaigns when optimising lead compound’s ADMET properties. Computational studies are appealing for virtually screening large number of drug candidates and often aim to identify their potential sites of metabolism (SOMs). Current SOM prediction tools are often drawing structure-reactivity relations from experimental data with few applying computationally expensive ab initio methods. Here we present an outline for creating a reactive ML-fitted force field for Cytochrome P450-catalysed reactions of drug-like molecules. A number of approximations will have to be made to collect enough DFT reference data and a potential specific to one or few simple compounds only will first have to be drafted. That said, ML force fields offer advantages of quantum mechanical accuracy, speed of classical force fields, and intrinsic ability to treat reactions and capture non-straightforward interactions of metal ions.
What data is really needed for the synthesis routes’ prediction and why data preparation is still a bottleneck? – Elena Herzog (Elsevier)
Prerequisites for AI/ML predictive modelling are mainly three: a powerful and scalable IT infrastructure designed to process large datasets, a large quantity of high-quality experimental data and proper descriptors that allow translation of the original, analog data into machine-readable data enabling preparation of an accurate AI/ML model with predictive functionality. In this regard, a key step of any AI/ML model generation is preparation and correct transformation of data, especially, if it comes from disparate sources where data models of data extraction, normalization and storage might differ. In the absence of standards on how chemical reactions data are extracted, annotated and stored, preparing the data for machine learning processes requires knowledge of what type of data are needed to answer a specific scientific question, how data have been indexed, unambiguous description of what has been extracted and what chemical descriptors were used to transform the extracted data from the literature into machine-readable data. The Entellect Reaction Workbench provides the IT infrastructure, tools and capability to work with multiple and diverse datasets for chemical and pharmacological predictive modelling to leverage AI potential in drug discovery. As a proof of concept, various reaction datasets were loaded into the Reaction Workbench in order to prepare a combined and de-duplicated dataset for subsequent use in predictive retrosynthesis model. At first sight, only a small overlap between the data sets was observed due to their underlying diverse reaction models. After careful analysis and identifying diversities in how reactions were extracted and recorded, three procedures of de-duplication and normalization were proposed prior to loading of the combined and cleaned dataset onto the platform which also increased the overlap between the various datasets. The procedures created will become part of the Entellect Reaction Workbench and help data scientists to transform, compare and de-duplicate disparate reactions datasets and to prepare them for various predictive modeling activities which will facilitate a speedy and effortless reaction data cleanup and preparation step.
A Platform for Automating Catalytic Chemical Synthesis to Understand a Complex Pd-Catalysed Reaction System Using Data Analysis, Mechanistic Studies and Reaction Optimisation – Christopher Horbaczewskyj (University of York)
C. S. Horbaczewskyja†, G. E. Clarkea‡, J. D. Firtha, J. T. W. Braya, L. A. Ledinghama, P. L. Martina, J. Toutaina, J. M. Lynama, J. M. Slatterya, J. Wilsona, R. A. Bourneb and I. J. S. Fairlamba* a Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK; b School of chemical and process engineering, University of Leeds, Leeds, LS2 9JT, UK; † chris.horbaczewskyj@york.ac.uk, ‡ gc627@york.ac.uk, * ian.fairlamb@york.ac.uk
Reaction automation has seen a boom throughout the past decade in which Chemists may now perform large numbers of reactions without the tedium of multiple reaction setups. The benefits of automation often outweigh the drawbacks i.e. optimisation acceleration, increased productivity, higher quality more robust data, amongst others. Reaction side products in the target molecule represent a safety regulatory concern. On the other hand, side products forming through potentially controllable reaction pathways may be exploitable in identifying new reaction methodologies. The work outlined employs a Chemspeed platform which allows full automation of reaction setup i.e. solid and liquid handling, temperature and stir control, the use of air sensitive reagents and at-line HPLC analysis. This platform has been used to perform and monitor a palladium cross-coupling of 2-bromo-N-phenylbenzamide where a plethora of products are created (Figure 1).
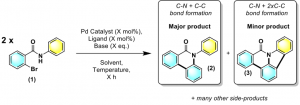
Figure 1 A complex Pd-catalysed reaction which affords a plethora of products.
Reaction automation of (1) focuses on product distribution where a single reaction forms over 10 products. A reaction screen of solvents, temperatures and times has been carried out using LC-MS for analysis. This data allowed interpretation using regression decision tree analysis and statistical pattern recognition methods, such as principal component analysis (PCA). The rich data set is being used to complement mechanistic understanding, by comparison with evidence gained by traditional methods along with other methods of automation i.e. a self-optimising continuous flow reactor.
Atom-To-Atom Mapping: A Benchmarking Study – Arkadii Lin (University of Strasbourg)
The atom-to-atom mapping (AAM) procedure is an important step of reaction data treatment allowing to identify a reaction center, i.e., an ensemble of atoms involved in formed, broken or modified chemical bonds. Despite the existence of various public/open source and commercial AAM tools, no exhaustive benchmarking study was performed so far. In this study, we compared some popular AAM tools (ChemAxon, Indigo, RDTool, CP-Solver) as well as their different combinations. These tools were applied to a set of 1052 manually curated and mapped reactions. In order to assess the efficiency of the studied tools, two parameters were recorded: (1) percentage of correctly mapped reactions (precision rate; PR) and (2) CPU time. Among studied tools, RDTool was found as the most accurate one (PR=79.75%), whereas it was very slow (2.7 s per reaction, on average). In contrast, ChemAxon was much faster (about 15 ms) but less efficient (PR = 75.29%). Other tools demonstrated similar to ChemAxon speed but lower PR (30.51% and 65.3% for Indigo and CP-Solver, respectively). In order to decrease the error rate, for a given reaction, we suggested choosing a tool providing with the smallest values of “Chemical Distance” calculated as the number of created, broken and transformed bonds. To reduce the number of erroneously mapped reactions and, hence, to increase the PR, a new rule-based strategy was also suggested.
Formation of artificial cells with distributed cores as multi-functional microreactors using 3D-printed microfluidics – Jin Li and David Barrow (Cardiff University)
The interest in chemically organised artificial cells has significantly increased in the last few decades. One of the key challenges in the creation of such miniaturised entities is to impart new functionalities to cell-like, soft architectures. This is typically determined by the spatiotemporal organisation of chemical reactions, which is underpinned by the compartmentalisation of various reagents in picolitre droplets, e.g. vesicles or coacervates. In our recent work, we demonstrate the formation of mobile artificial cells, incorporating droplet networks and nanomaterials, using multi-material 3D-printed microfluidic devices. This approach not only enables the precise control of chemical compartmentalisation, but also shows the ability to scale up the production of artificial cells, for further research on their group-level behaviours. We are currently working on the orchestration of the liquid “cores” within the artificial cells using computer-assisted manners, that the distribution of different cores determines the pathway of internal chemical-mediated information transfer.
Reaxys Predictive Retrosynthesis (PAI): rewiring chemistry and redesigning synthetic routes – Elliott Parris (Elsevier)
Abhinav Kumar, Jurgen Swienty-Busch, Thomas Bottjer, Ivan Krstic.
Synthetic organic chemistry is one of the cornerstones of the drug discovery process and is crucial for the rapid navigation of increasingly complex druggable chemical space. It is also critical in determining discovery cycle times and development speeds associated with drug candidates (Lowering et al. J. Med. Chem., 2009 & Nadin et al. Angew. Chem., Int. Ed., 2012). The knowledge and experience of researchers has always been the key to combining chemical reactions into successful synthetic schemes. However, chemical synthesis and route design is still a significant challenge. Merck quotes that 55% of the time, a benchmarked reaction fails (Science 02, 2015). Therefore, a radical and innovative step-change in synthesis is needed. Computer-aided retrosynthesis would be a valuable tool but, at present, it is slow and provides results of unsatisfactory quality. Further to this, software that devises effective schemes for synthetic chemistry has depended on the input of rules from researchers. Waller and Segler et al. (Nature, March 2018) in collaboration with Elsevier have developed a ‘deep learning’ computer program that produces blueprints for the sequences of reactions needed to create small organic molecules, such as drug compounds. This artificial-intelligence tool has digested nearly every reaction ever performed (> 15 million) and has the potential to transform the way how synthetic chemists work in the future. Segler & Waller et al. tested the synthetic routes that the program threw up in a double-blind trial with 45 organic chemists from two institutes in China and Germany and the routes have proven scientifically sound and robust. Increasing the success rate in synthetic chemistry would have a huge benefit in terms of speed and efficiency on drug-discovery projects, as well as cost reduction. Reaxys-PAI Predictive Retrosynthesis solution developed in collaboration between Elsevier and Waller and Segler et al. is a first-in-class & best-in-class solution combining unparalleled Reaxys content with AI & ML technologies. It deploys next-generation technologies to augment chemical synthesis and chemist knowledge helping drive innovation, time and cost savings. The Reaxys predictive retrosynthesis tool will be an invaluable assistant for the chemist who wants to make molecules as quickly and as confidently as possible.
Affiliations: Elsevier Information Systems GmbH, Frankfurt, Germany
*Reaxys is a trademark of Elsevier Limited and PAI is Pending AI Pvt. Ltd.
Computational Methods to Identify Novel Medicinal Chemistry Relevant Heterocycles – Fergus Preston (Drug Discovery Unit, University of Dundee)
The necessity for pharmaceutical companies, and indeed academia, to efficiently screen libraries of compounds to generate hits is an ever challenging endeavour. It is currently estimated that the chemical space, which encompasses synthesised and un-synthesised molecules, is 100 million compounds. (1) This number of possible compounds increases to as large as 1060 molecules when all of Lipinski’s structural rules are taken into consideration. (2,3) However despite these vast available chemical libraries, many of the compounds found within are flat, planar, lipophilic compounds that lack sufficient chemical diversity to be classified as distinctly ‘different’ from one another and therefore the likelihood of probing new chemical space is very low. Filtering a set of 26 million compounds (4) through pipeline pilot and cross-referencing against Reaxys, has identified 1.7 million heteroaromatic compounds that are undocumented in the literature. Using protocols that remove any toxic structural alert from the molecule and then applying a computational algorithm that assesses the compounds’ synthetic accessibility and provides a numerical, quantitative value based on molecular complexity (size, fused rings, stereocentres) as well as fragment complexity based on the frequency of appearance in datasets of known synthesised molecules, filtered the set to 700,000 compounds. Using a prototype software (5) we were able to score the compounds for elaboratability – that being fragments that have more hits particularly across different classes of chemical reaction and thus, potentially more readily elaborated. Lastly, using computational assessment of organic space (COAS) we were able to filter the set down by ring size to identify synthesisable novel heterocycles.
- Kirkpatrick, P.; Ellis, C., Nature 2004, 432, 823-865.
- Bohacek, R. S.; McMartin, C.; Guida, W. C., Med. Res. Rev. 1996, 16, 3-50.
- Lipinski, C. A.; Lombardo, F.; Domivy, B. W.; Feeney, P. J., Adv. Drug. Delivery. Rev 1997, 23, 3-25.
- Ruddigkeit, L.; van Deursen, R.; Blum, L. C.; Reymond, J. L., J. Chem. Inf. Model 2012, (52), 2864-2875.
- Mayfield, J., NextMove Software, Pistachio.
A comprehensive modeling methodology for the development of QSPR models for kinetic characteristics of chemical reactions – Assima Rakhimbekova (Kazan Federal University)
Reactions is one of the main objects of interest in chemistry. It would be great to predict whether the reaction will go, how quickly it will go, under what conditions and so on. In the presentation, we demonstrate our recently developed techniques and tricks for modelling kinetic and thermodynamic characteristics of reaction. Descriptors. The structures of reactions can be encoded in reaction feature vectors using difference between count-based fingerprints of products and reactants [1-3], extended SiRMS mixture representation approach [4], as well as concatenation of reactants and products descriptors. The novel methodology of reaction representation based on Condensed Graph of Reaction approach can be used to calculate descriptor vector [5] or apply graph-convolutional neural network. Comparison of these approaches of reactions representation will be given. Condition influence. Modelling of the properties of chemical reactions requires not only the structures of reactions but also conditions of reactions (T, solvent, catalyst and so on). The preparation of a descriptor vector encoding a condition of reaction will be discussed. Model validation. Since reaction description includes not only the structure of components but also conditions, rigorous validation of the model should take it into account. Two strategies of model validation: «transformation-out» and «solvent-out» – will be described. Comparison of these validation strategies with simple «reaction-out» validation will be given. Applicability domain. QSPR models are not universal, consequently, the quality of object prediction highly depends on its similarity to the training set [6-7]. AD definition approaches suitable for models predicting reaction characteristics was proposed and benchmarked. A large number of different AD definition approaches extensively used in QSAR/QSPR studies, their modifications, as well as novel approaches designed by us for reactions was used in comparison and the best approaches according to the number of criteria were selected. Data. The models predicting the reaction rate of bimolecular substitution [8], elimination [9], Diels-Alder reactions [10] as well as tautomeric equilibrium constants [11] were built and validated.
Acknowledgements. Research was supported by the Russian Science Foundation grant 19-73-10137.
References:
- Schneider, N.; Lowe, D. M.; Sayle, R. A.; Landrum, G. A. J. Chem. Inf. Model. 2015, 55, 39–53.
- Carhart R.E.; Smith D.H.; Venkataraghavan R. J. Chem. Inf. Comput. Sci. 1985, 25 (2), 64–73.
- Rogers D.; Hahn M. J. Chem. Inf. Model. 2010, 50 (5), 742–754.
- Polishchuk P., Madzhidov T., ·Gimadiev T., · Bodrov A.,· Nugmanov R., · Varnek A. J. Comput. Aided. Mol. Des. 2017, 31, 829–839.
- Varnek, A.; Fourches, D.; Hoonakker, F.; Solov’ev, V. P.; Solov’ev, V. P.; Solov’ev, V. P. J. Comput. Aided. Mol. Des. 2005, 19, 693–703.
- Jaworska J.; Nikolova-Jeliazkova N.; Aldenberg T. Altern Lab Anim. 2005, 33, 5, 445-459.
- Tetko I.V.; Sushko I.; Pandey A.K.; Zhu H.; Tropsha A.; Papa E.; Oberg T.; Todeschini R.; Fourches D.; Varnek A. J. Chem, Inf. Model. 2008, 48, 9, 1733-1746.
- Gimadiev T.; Madzhidov T.; Tetko I.; Nugmanov R.; Casciuc I.; Klimchuk O.; Bodrov A.; Polishchuk P.; Antipin I.; Varnek A. J. Mol. Inf. 2018, 38, 4, 1800104-1800119.
- Madzhidov T.I.; Bodrov A.V.; Gimadiev T.R.; Nugmanov R.I.; Antipin I.S.; Varnek A. J. Struct. Chem. 2015, vol. 56, no 7. pp. 1227–1234.
- Madzhidov T.I.; Gimadiev T.R.; Malakhova D.A.; Nugmanov R.I.; Baskin I.I.; Antipin I.S.; Varnek A. Journal of Structural Chemistry, 2017, vol. 58, no. 4, pp. 685-691.
- Gimadiev T.; Madzhidov T.; Nugmanov R.; Baskin I;, Antipin I.; Varnek A. Journal of Computer-Aided Molecular Design. 2018, 32, 3, 401-414.
Catalyst Design via Machine Learning – Stamatia Zavitsanou (Oxford University)
Stamatia Zavitsanou, Tom Watts, Veronique Gouverneur, Fernanda Duarte, Department of Organic Chemistry, University of Oxford.
The production of medicines, fuels, foods, and fertilizers rely on catalysts. However, despite their importance, the question about how exactly chemists can develop new catalysts, without depending on laborious trial-and-error, remains. Experimentally modifying catalysts at different positions is a laborious and time-consuming process. To this end, computational methods provide a complementary approach, guiding the design of more efficient catalysts at an early stage in the process of discovery. In this project, we aim to expand the methodology of hydrogen-bonding phase-transfer (HB-PT) catalysis, as reported by the Gouverneur group1, 2, via computational studies. To achieve this goal, we use density functional theory (DFT) and machine learning (ML). We develop a linear regression (LR) model trained on multidimensional chemical data. It can be used to predict the performance of an HB-PT reaction and to infer underlying reactivity of nucleophilic fluoride for both episulfonium and aziridinium substrates. We show that a set of simple atomic and molecular descriptors, which can be automatically extracted from currently used electronic structure packages, can serve as an input for a LR model to predict reactivity. Ultimately, we wish to implement an automated workflow to streamline the computational design of new catalytic scaffolds. Our intention is to change the way chemists select and optimize catalysts from an empirical to a mathematically guided approach.
References:
- G. Pupo, F. Ibba, D. M. Ascough, A. C. Vicini, P. Ricci, K. E. Christensen, L. Pfeifer, J. R. Morphy, J. M. Brown, R. S. Paton and V. Gouverneur, Science, (2018), 360, 638-642.
- G. Pupo, A. C. Vicini, D. M. Ascough, F. Ibba, K. E. Christensen, A. L. Thompson, J. M. Brown, R. S. Paton and V. Gouverneur, JACS, (2019), 141, 2878-2883.